背景
打分函数广泛应用于基于结构的计算辅助药物设计,其通过定量化评估药-靶的相互作用为药物研发中的药效评估提供理论依据[1-5],提高活性化合物甄别的效率。定量评估药物与靶标蛋白的相互作用通常分为两步,一步是对接处理(docking process),主要指构象搜索,找出潜在的binding pose;另一步是打分处理(scoring process),通常指打分,预测药-靶结合力。大部分打分函数并不基于完整的物理模型而作近似处理,因此往往不严格遵循多体扩展理论,守恒定律,对称不变性等,甚至有些基于知识的打分函数的表达式完全不含物理意义。事实上,作为应用于药物高通量筛选场景下的一种工具,大部分打分函数以效率为中心,通过近似的手段追求精度与效率的平衡[6]。
打分函数分类
从上世纪的90年代起,已报道的打分函数已经有超过一百种。先前已有一些工作综述过打分函数的分类,比如将打分函数分为基于力场的,基于经验的,基于知识的三种类型。近年来随着这个领域的研究不断深化,一些新的技术比如机器学习的引入带来的描述符的引入,加上基于经验的打分函数的表达式与基于力场的表达式的相似性,原有的打分函数分类描述会让初涉此领域的研究人员感到迷惑,因此王任小课题组在2015年结合打分函数研究的进展,对打分函数做出了新的分类的阐述[6]。其将打分函数大致进行了以下四类的阐述:分别是基于物理(力场)的打分函数,基于经验(回归)的打分函数,基于平均力势(知识)的打分函数,基于描述符(机器学习)的打分函数。下面通过推导原理与代表性的函数表达式对四种打分函数作进一步的介绍。
基于物理(physics-based)的打分函数
按传统划分此类打分函数是基于力场的打分函数。
能量函数推导形式
基于明确物理意义的物理模型推导。能量函数家族由基于实验数据拟合的力场函数,基于第一性原理的量化计算函数,基于连续介质的溶剂模型等组成。
下面举一个典型的表达式例子:

其中,能量由范德华力项,静电力项,氢键项,去溶剂化项组成。
常见的基于物理的打分函数
COMBINE, GoldScore,MedusaScore,Linear Interaction Energy (LIE),Linear Response Approximation (LRA),MM-PBSA/GBSA。
优点
能够很好利用现代力场开发,量化计算,溶剂化模型等理论模拟计算方法的研究进展。
缺点
由于构成能量函数的每个单项的内在偏差,实际计算的结果可能会远远大于实验观察值的偏差(实验观察值波动的典型范围在3~18kcal/mol),需要用经验参数拟合实验观察值与最后的计算结果从而得到最终函数形式。
基于回归(regression-based)的打分函数
按传统划分此类打分函数是基于经验的打分函数。
能量函数推导原理
经验打分函数通过求和一些权重化的独立项的能量贡献来计算蛋白质-配体结合的结合力,每个项代表蛋白质-配体结合中一个重要的能量因子。基于经验的打分函数的每一个独立项通过回归的方法进行权重因子(在函数的每个独立项前设置参数)的计算。
下面以ChemScore的表达式为例子作进一步解释:

其中,“S”代表rewardingscores,分别对应氢键,金属离子配位,亲脂的rewarding scores。“P”代表penalty rewards,分别对应冻结旋转键,内部应变能,空间碰撞。如果需要共价对接或限制对接,可能会受到额外处罚。
常见的基于回归的打分函数
PLP, ChemScore, X-Score,GlideScore。
优点
基于经验的打分函数在第一地点(配体-靶标蛋白结合点)进行校准,从拟合的角度看,拟合操作本身所保留信息相对于其他打分函数会更加完整。另外与基于物理的打分函数的单一项对比,相应的基于经验的单一项的物理意义更加直观(比如基于物理的打分函数的氢键项是通过二体或多体力场函数来表达,而基于经验的氢键项则没有这些表达式。)。采用直观函数形式的结果实际上是双刃的。一方面,它是一个技术优势,因为它便于实现任何合理的想法。这样一个例子是GlideScore-XP,它可以说是目前最复杂的经验评分函数。GlideScore-XP的设计强调通过奖励或惩罚某些相互作用模式来识别蛋白质结合位点的多样性。特别感兴趣的是将氢键分为中性-中性、中性-带电和带电类型,除了考虑蛋白质和配体之间的疏水接触外,还使用单独术语解释“疏水外壳”。添加或删除单个术语的便利性也使得为某些分子系统开发定制的评分函数以获得更好的性能成为可能。
缺点
1 某些相互作用模式的缺失。采用直观的函数形式增加了这些方法的经验性质。经验评分函数仅包括常见的蛋白质-配体相互作用模式。不太常见的相互作用模式,尽管很强,而且特殊的,如阳离子-π相互作用,通常被忽略,因为它们在回归分析中并不显著。或者,如果某个因素不能被人以直接的方式解释,例如熵因子,它也不可能被包括在内。因此,在经验评分函数的框架内建立对蛋白质-配体结合中所有可能因素的全面和一致的描述是相当困难的。
2 因其是基于对实验数据的直接拟合,所以数据集本身存在的一些误差,比如不同方法、不同来源的实验值造成的误差,对结果的影响会更加明显。
注意
经验打分函数和基于物理的方法之间的界限往往不像人们想象的那样明显。事实上,两者都将结合自由能的蛋白质-配体分解成单独的能量项。此外,基于物理的方法可以引入经验参数来调和其能量项的贡献,就像经验打分函数一样。然而,将经验打分函数与基于物理的方法分开来讨论仍然是有帮助的。它们之间的主要区别在于,基于物理的方法从其他成熟的模型中借用了完整的理论框架,包括能量函数形式和相关参数;而经验打分函数通常采用灵活、直观的函数形式,是从头设计组成的。
基于知识(knowledge-based)的打分函数
也有人将此类打分函数称为平均力势(potential of mean force)打分函数。
能量函数推导原理
基于知识打分函数通过求和蛋白质-配体对统计势函数来计算蛋白质-配体结合的结合力(fitness),而距离依赖的势函数ωij(r)由反玻尔兹曼分析推导。

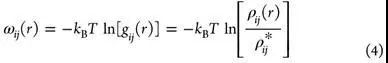
其中ρij(r)是在距离r处,原子对i-j的数值密度,ρij*是相同原子对在原子间相互作用假定为零的参考状态下的数值密度。通过这种方法,假设成对接触的发生频率是其对蛋白质-配体结合的能量贡献的一种度量。如果特定的成对接触比参考状态(即随机分布)下更频繁发生,,则表示给定原子对之间的能量是有利的相互作用;如果不经常发生,则表示不利的相互作用。为了获得所需的成对电位,标准方法是使用一组来自PDB的蛋白质-配体复合结构作为训练集,即“知识库”。蛋白质侧和配体侧的原子根据其分子环境被分类为许多简并原子类型。然后,根据在公式4的训练集中观察到的原子对的出现频率,推导出每个可能的原子对的距离依赖势。
常见的基于知识的打分函数
Muegge’s PMF, DrugScore,IT-Score,KECSA。
优点
作为一个起源于液体统计力学分析的基本思想,逆玻尔兹曼分析可以有效地将原子间距离的柱状图转换为平均力势。其优点是:1 概念和计算简单。2 不受实验数据约束。
缺点
蛋白质和配体都不是液体中原子的随机组合。相反,分子中的原子被共价键按一定的顺序约束。因此,式4中考虑的参考状态不符合真实参考状态的定义。由于同样的原因,不同原子对的出现并不是完全独立的。一些分析指出,在实际的蛋白质-配体复合结构中,某些原子对的出现频率不应假定为Boltzmann分布[7-8]。尽管由式4得出的统计势函数通常被认为是平均力势函数的近似值。这一解释实际上并不可靠。
基于描述符(descriptor-based)的打分函数
也有人将此类打分函数称为机器学习(Machine Learning)打分函数。
能量函数推导原理
首先构建具有一定物理意义(虽然有一定的模糊性)的描述符,比如描述特殊相互作用的描述符(静电相互作用、氢键或芳香堆叠)、几何描述符(表面或形状特性)和传统的基于配体的描述符(分子量、可旋转单键数等)等,然后建立机器学习计算模型,继而由机器学习算法推导能量函数,通常函数形式为非线性的灵活多变的表达式,没有固定的形式。
常见的基于描述符的打分函数
NNScore, RF-Score, SFCscore,and ID-Score。
优点
与上面的其他打分函数相比,基于描述符(机器学习)的打分函数所计算的结果与实验数据的相关性更好。
缺点
当前大部分基于机器学习的打分函数开发者将此模型作为黑匣子来使用,打分函数的形式不具有明显可解释的物理意义,没有明确或者充分考虑溶剂效应,熵效应,多体效应。
附:CASF-2016采用的主流打分函数分类汇总
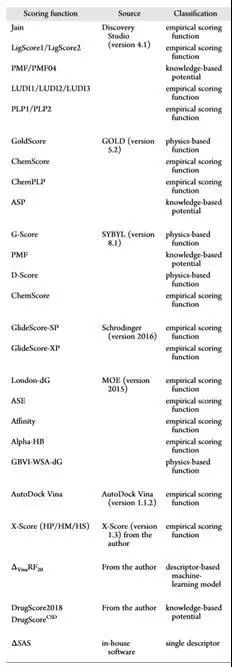
图 1 CASF-2016采用的主流打分函数分类汇总[9]
评价打分函数的Metrics
打分能力(Scoring Power)
不同配体,不同靶标蛋白。打分能力通过计算打分函数产生的binding score与实验binding data(通常是logKa)的线性相关性来进行评估。常用的定量化indicator是Pearson’s correlation coefficient(R),Standard Deviation(SD)可以作为辅助的indicator。
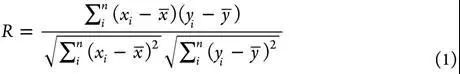
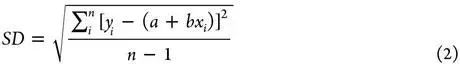
其中xi 是第i个复合物根据打分函数计算出来的结合力,yi 是第i个复合物的实验结合力,a是回归线的截距,b是回归线的斜率。
排序能力(Ranking Power)
不同配体,同一靶标蛋白。在给定不同配体的精确binding pose的前提下,对同一个靶标蛋白结合不同配体进行排序以找出最佳的配体-蛋白组合。常用的定量化indicators有Spearman’s rank correlationcoefficient(ρ),Kendall’s rankcorrelation coefficient(τ),PredictiveIndex(PI)。

其中,rxi是第i个复合物根据打分函数计算得到的排序,yi是第i个复合物根据实验亲和力得到的排序,n是排序的总样本数。

其中,Pconcord 代表排序一致的对数,Pdiscord代表排序不一致的对数,一对用(xi ,yi)表示,如果x的排序与y的排序一致(比如xi >x j 且 yi >yj, or x i <x j 且yi<y j),则认为排序一致,如果(i.e.,xi >xj 且yi <yj, or xi<x j 且 yi >yj).则认为是不一致。T是x中的捆绑数(xi=x j ),U是y中的捆绑数(yi =yj)。如果 xi=x j 且yi=yj 同时发生,一对则不计入T和U。

这里,Wij是配体i和j结合数据之间的差值,即Wij=abs(Ej−Ei)。Cij表示实验结合数据(Ei和Ej)和预测结合分数(Pi和Pj)的排序顺序是否一致。也就是说,如果(Ej−Ei)/(Pj−Pi)>0,Cij=1;如果(Ej−Ei)/(Pj−Pi)<0,Cij =−1;如果(Ej−Ei)/(pj−pi)=0,Cij=0。
上面描述的所有三个指标都在-1到+1之间,其中+1表示一个完美的排名,而-1表示一个完全相反的排名。
对接能力(DockingPower)
同一配体,同一蛋白,不同binding poses。在给定天然配体binding pose(native binding pose)及不同的binding poses(包含native binding pose和一定数量的decoy)的前提下,对同一个靶标的对接进行打分,找出native binding pose。理想情况下,native binding pose应与排名靠前的pose相同。利用所测试的打分函数对每个复合体的诱饵集(decoy set)进行打分。Native binding pose和该打分函数选择的最佳打分binding pose之间的RMSD值是使用Allen等人10开发的匈牙利算法计算的,如果RMSD差异低于预设的临界值(例如2.0埃),则该复合体被标记为成功的binding。对整个测试集进行分析,然后将总体成功率作为对接能力的定量指标。
CASF-2016对接能力试验又增加了一项新的进展,称之为“结合漏斗分析”,其目的是使天然配体结合位置对应于正确结合能面上的最低值。期望结合能面在最低值附近具有漏斗状形状,其中具有较小RMSD值的配体结合位与较低的结合能相关联。如果是这样的话,分子对接过程中的构象采样将更有效地引导到最终目的地。在CASF2016中,在decoy set的不同binding poses的RMSD值和他们计算出的binding scores之间的Spearman排列相关系数被用来作为定量化这种关系的indicator。对于正在测试的打分函数,记录在测试集中所有蛋白质-配体复合物上获得的Spearman排列相关系数的平均值。此外,为了探索结合漏斗的范围,通过考虑某个RMSD窗口中的诱饵结合姿态,计算出每个复合体的Spearman排列相关系数。总共考虑了九个RMSD窗口,包括[0−2埃_]、[0−3埃]、[0−4埃]、[0−5埃]、[0−6埃]、[0−7埃]、[0−8埃]、[0−9埃]和[0−10埃]。
筛选能力(Screen Power)
指打分函数在一组随机分子中识别与给定目标蛋白真正结合的能力。在CASF-2016中,筛选能力基本上在交叉对接试验中进行了评估。试验组共有57种靶蛋白。每个靶蛋白包含五个复合物,因此,每个靶蛋白都有五个已知配体。这些已知的配体被视为阳性,其他285−5=280个配体被视为阴性。对于目标蛋白,测试的打分函数被应用于所有285个配体的打分。在CASF-2016中,共为每对蛋白质-配体制备了100个具有代表性的配体结合位点。将所测试的打分函数应用于计算所有这些结合位置,其中最佳结合分数记录为该蛋白质-配体对的预测结合分数。然后,所有285个配体按结合分数降序排列。打分函数的筛选能力是通过它把真正的结合排在首位的程度来评估的。
筛选能力的第一个定量指标是在测试集中所有57个靶蛋白的1%、5%或10%的top-ranked配体中识别最高效率结合物的成功率。第二个指标是富集因子(EF),计算如下:

其中NTBα 是在α下实际结合的数目,α是最高排行的比例数,NTBtotal是给定一个靶标下总的真实的结合数目。
评价打分函数的benchmark数据集
综合性数据库:PDBBind [11], http://www.pdbbind-cn.org/
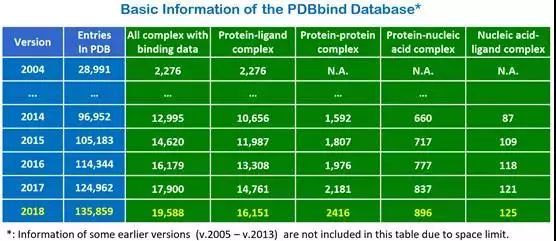
图 2PDBbind数据库基本信息
数据库提供:
1 结合力数据
2 经过加工的结构文件
3 基于网页的展示和分析工具
测试数据集:
CASF-2013 12, http://www.pdbbind-cn.org/casf.asp
图 3CASF-2013数据集目录总览
其他测试数据集:
CSAR_HiQ_NRC_set (Dunbar et al., 2013) ,http://www.csardock.org/
主流打分函数对比评估
数据集:CASF-2016
具体结果如下:以下所有图片所结果按评价的指标由大到小排列,对于每一项能力的单独评价,以ΔSAS为参考打分模型,具体分析请参考文献[9]。
打分能力
评价指标:Pearson Correlation Coefficient
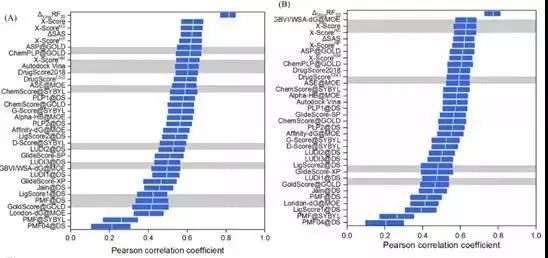
图 4CASF-2016对比评价—打分能力
排序能力
评价指标:Spearman Correlation Coefficient
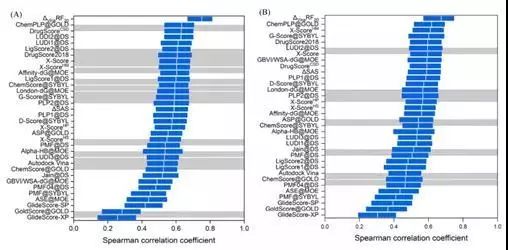
图 5CASF-2016对比评价—排序能力
对接能力
评价指标:成功率。
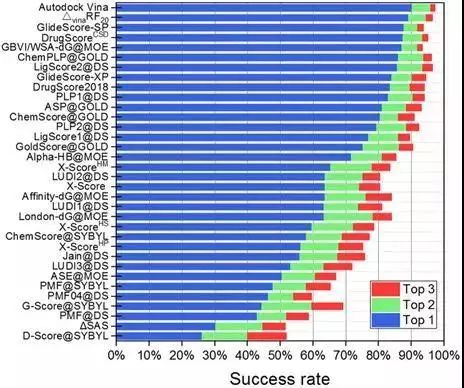
图 6CASF-2016对比评价—对接能力
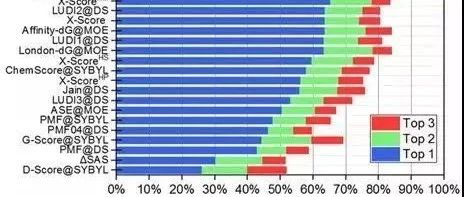
筛选能力
评价指标:富集因子(enrichment factor)。
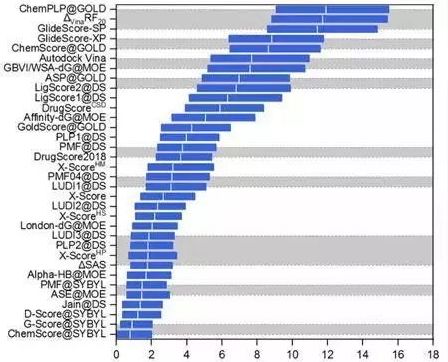
图 7CASF-2016对比评价—筛选能力
以上评估结果关键对比信息基本上一目了然,这里不按图一一解释,若需进一步了解更加详细的打分结果阐述,可以参考文献[9]。
作为示范,CASF-2016评估了25个打分函数。这些打分函数来自多个主流分子模型软件,也有一些来自学术团体。结果表明,被测打分函数的对接能力是最有希望的,其中一些打分函数能够达到70%以上的成功率。然而,即使是排名靠前的打分函数(除ΔVinaRF20外),在打分能力和排名能力测试中也只能产生0.60左右的相关系数。筛选能力是打分函数最薄弱的方面,排名靠前的打分函数产生的成功率约为40%。对接能力与得分/排名能力之间没有明显的相关性。然而,得分能力和排名能力有一定的相关性,对接能力和筛选能力也有一定的相关性。此外,大多数打分函数仅在一个或两个方面相对成功,某些打分函数在所有方面表现出更为平衡的性能,如ΔVinaRF20和ChemPLP@GOLD。此外,在CASF-2016和CASF-2013的结果中观察到了相似的趋势,这表明测试数据集产生了稳健的评估结果。
虽然CASF-2016打分函数对比评估仅测试了有限数量的打分函数,但仍可以观察到近年来开发的打分函数优于旧的打分函数。例如,在对接功率和筛选功率测试中排名最高的打分函数包括ΔVinaRF20, AutoDock Vina, ChemPLP@GOLD, GlideScore-SP, 和 GlideScore-XP,所有这些函数都是相对较新的。这证实了打分函数开发领域作为一个整体正在向前发展。特别是,借助机器学习技术开发的打分函数代表了一种新的潮流。一些机器学习模型已经在其他研究人员的CASF-2013基准测试中进行了改进,证明了其优于传统打分函数。作为CASF2016中这类打分函数的选定代表,ΔVinaRF20在打分能力和排序能力测试中占有明显的领先优势。它的对接能力和筛选能力测试也在前三。尽管ΔVinaRF20在研究中产生的结果需要谨慎对待,但还是应该相信机器学习技术将加速打分函数的发展。
参考文献:
(1) Muegge, I.; Rarey, M. Small moleculedocking and scoring. In Reviews in Computational Chemistry; Lipkowitz, K. B.,Boyd, D. B., Eds.; Wiley-VCH Inc.: New Jersey, 2001; Vol. 17, pp 1−60. (2) Böhm, H. J.; Stahl, M. The use of scoringfunctions in drug discovery applications. In Reviews in ComputationalChemistry; Lipkowitz, K. B.; Boyd, D. B., Eds.; Wiley-VCH Inc.: New Jersey,2002; Vol. 18, pp 41−88. (3) Schulz-Gasch, T.;Stahl, M. Scoring functions for protein-ligand interactions: a criticalperspective. Drug Discovery Today Technol. 2004, 1, 231−239. (4) Leach, A. R.; Shoichet, B. K.; Peishoff, C. E. Predictionof Protein-Ligand Interactions. Docking and Scoring: Successes and Gaps. J.Med. Chem. 2006, 49, 5851−5855.
(5) Rajamani, R.; Good, A. C. Ranking posesin structure-based lead discovery and optimization: Current trends in scoringfunction development. Curr. Opin. Drug Discovery Develop. 2007, 10 (3), 308−315.
(6) Minyi Su, Qifan Yang, Yu Du, Guoqin Feng, Zhihai Liu, Yan Li,and Renxiao Wang
Journal of Chemical Information and Modeling 2019 59 (2), 895-913.
(7) Ben-Naim, A. Statistical Potentials Extracted from ProteinStructures: Are these Meaningful Potentials? J. Chem. Phys. 1997, 107, 3698−3706.
(8) Thomas, P. D.; Dill, K. Statistical Potentials Extracted fromProtein Structures: how accurate are they? J. Mol. Biol. 1996, 257, 457−469.
(9) Minyi Su,Qifan Yang, Yu Du, Guoqin Feng, Zhihai Liu, Yan Li, andRenxiao Wang. Comparative Assessment of Scoring Functions: The CASF-2016 Update.J. Chem. Inf. Model. 2019, 59, 895−913
(10) Allen, W. J.; Rizzo, R. C. Implementation of the HungarianAlgorithm to Account for Ligand Symmetry and Similarity in Structure BasedDesign. J. Chem. Inf. Model. 2014, 54, 518−529.
(11) Zhihai Liu, Yan Li, Li Han, Jie Li, Jie Liu, Zhixiong Zhao, WeiNie, Yuchen Liu and Renxiao Wang, “PDB-wide collection of binding data: current status of the PDBbind database”,Bioinformatics, 2015, 31 (3): 405-412.
(12) Yan Li, Minyi Su, Zhihai Liu, Jie Li, Jie Liu, Li Han, RenxiaoWang *, “Assessing Protein-Ligand Interaction Scoring Functions with the CASF-2013Benchmark”, Nature Protocols, 2018, Vol. 3(4): pp666-680.(CASF-2013)
转载自QMCLab:打分函数分类及评价
