引言
AI为化合物的属性预测,药物设计以及分子逆合成提供了有效的解决方案,来自赛诺菲的Christoph Grebner在JMC专刊Artificial Intelligence in Drug Discovery中报道了基于AI的从头分子设计研究结果。

自动化分子从头设计已经被研究了大约20年,尽管许多工具已经被建立起来,许多成功的案例也已经被知晓。分子自动化从头设计仍然没有被普及开来。其中的部分原因可能是由于设计的分子很难被合成出来。本文介绍了基于强化学习来进行化学空间的探索的方案;同时,也对相关的评分方法进行了研究;最后,在对典型药物研发项目的回顾性研究中,作者研究了如何生成项目进展所需的相关分子,以及如何定制方案以满足分子合成和优化的相关需求。
生成性神经网络被训练以用于从头设计具有指定属性的化合物。训练过程主要包括两个步骤。首先,使用大型化学数据库来生成模型,该模型将会学习如何生成正确化学结构。在第二步中,使用RL训练这些模型,以便可以将生成的化学结构放到指定的化学特性空间中。
(1)生成模型,使用RNN(循环神经网络)来生成新型化学分子结构,这样的训练是基于一个非常庞大的分子数据库。RNN会学习SMILES的语法规则-SMILE是一种描述化学分子的简单线性语言。RL(增强学习)被用于指导RNN如何生成具有指定属性的可用分子。首先训练出的神经网络模型被称之为“prior”。

图 1. 强化学习路线图
来源:JMC
(2)化学空间,三个不同的数目庞大的化学数据库被用于训练prior。其中ChEMBL(version 24, ~1.45 million molecules),Sanofi(~3.37 million molecules)代表的生物活性分子,而Enamine(~5.36 million molecules)代表的是虚拟可及性分子。使用SMILES来展示这些分子,互变异构体暂时不被考虑在内。同时为了探索一些化学亚结构的遗失会对模型有什么影响,训练集移除了一些特定亚结构,形成了几个新的化学亚空间用于训练。A: No amides ;B: No amidines ;C: No guanidines;D: No benzene;E: No five-membered rings。
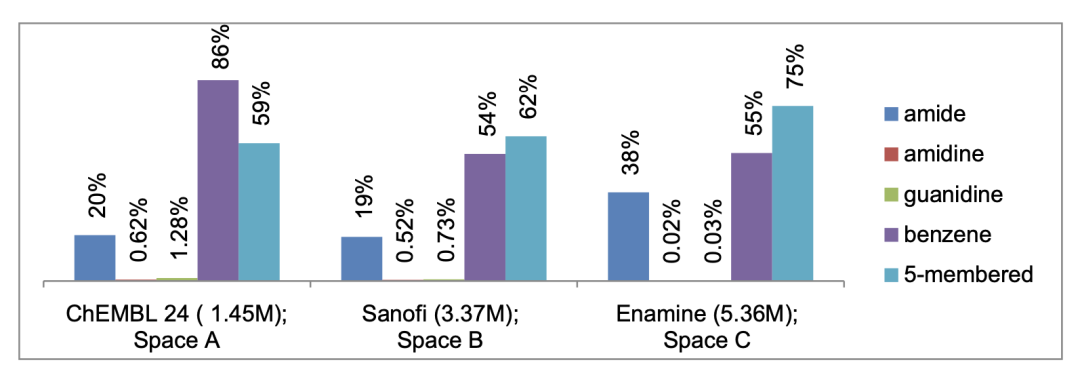
图 2.不同训练数据集的组成
来源:JMC
(3)打分函数,Prior 可以被用于生成化学结构,但是对于先导化合物的合成以及优化而言,需要去生成一些在特定范围内的化学分子。作者通过各种打分(包括化学相似性和各种基于QSAR的生物活性模型)将新分子的生成导向了所需的化学属性空间。被使用的打分函数:a:2D相似性(ECFP6 分子指纹);b:3D相似性(ROCS-3D);c: 2D-QSAR模型(基于Cubist regression trees);d:2D-QSAR-model (基于GraphConvolutions);e:组合打分函数

总结
AI在目前的生活中扮演着重要的角色,在药物发现中AI已经被应用了数年,并为相关的任务(如属性预测,分子设计和化学逆合成)提供了有潜力的建议。这些活动将加快会对药物相关分子的搜索,并有助于缩短工业界和学术界的药物研发周期。本文研究了人工智能分子从头设计的几个实际应用。为此,作者使用强化学习方法进行分子从头设计,评估了不同化学空间作为输入训练集,以及不同评分函数和组合对最后结果的影响。
这些结果表明,在研究中,可以设计一套定制的方案来助力药物研发相关工作。如果是想要生成相似化合物,在强化学习中使用2D方法进行打分可能有用,但显然会产生更为保守的结果。另一方面,如果目标是识别新的先导化合物,则单独或组合使用3D-shape和QSAR模型来对RL进行评分,可以获得更好的效果。因此,将更复杂的方法(例如3D-shape或QSAR模型)集成到打分中,可以对化学空间进行比较无偏的采样,从而提供新颖的思路。
参考文献
Grebner, C., et al., Automated De Novo Design in Medicinal Chemistry: Which Types of Chemistry Does a Generative Neural Network Learn? Journal of Medicinal Chemistry, 2020.