背景
新药研发通常需要耗费大量的资源,时间跨度10-20年,费用大约5亿美元到26亿美元。有望通过人工智来促进先导化合物发现来加速药物研发并降低成本。Discoidin domain receptor 1 (DDR1)是一种由胶原激活的促炎酪氨酸激酶受体,在上皮细胞中表达并参与纤维化。为了寻找到潜在的DDR1抑制剂,研究人员们开发了一种机器学习算法,用于设计新药分子。药物研发人工智能公司Insilico Medicine与药明康德联合在Nature Biotechnology报道了基于人工智能设计DDR1抑制剂的成功案例,并开源了相关代码。
基于深度学习的DDR1抑制剂发现流程
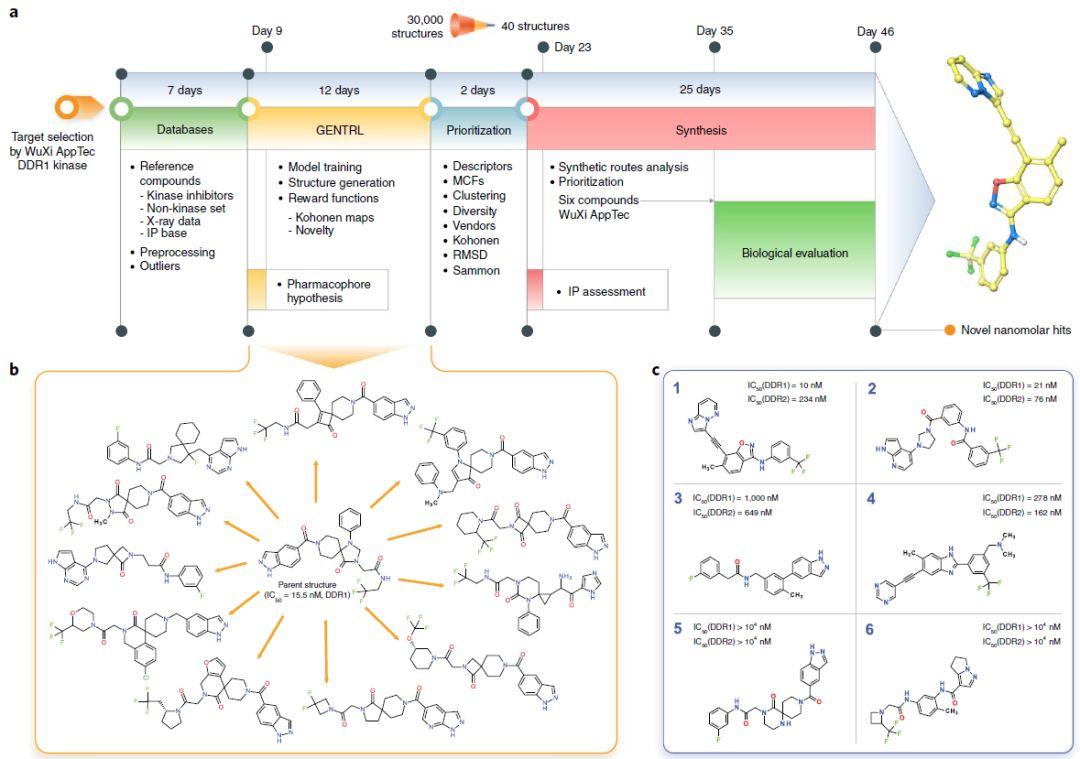
从靶点选择到活性分子筛选和验证,总时长耗费46天,信息收集以及数据整理7天,建立深度学习模型12天,优化2天,合成以及动物实验25天。

图1. 基于深度学习的DDR1抑制剂发现的工作流
图片来源Nature
首先,使用六个数据集来构建模型:(1)来自ZINC数据集的抑制剂,(2)已知的DDR1激酶抑制剂,(3)常用激酶抑制剂(阳性组),(4)非激酶抑制剂作用分子(阴性组),(5)制药公司的生物活性分子的专利数据(6)DDR1抑制剂的三维(3D)结构。在过滤的ZINC数据库上预训练GENTRL(预训练),然后在DDR1和常见的激酶抑制剂数据集上进行训练。随后进入强化学习阶段。初期获得了30,000个输出结构后过滤除去带有结构警报或反应基团的分子,并通过聚类和多样性分析减少分子的化学空间维度。然后使用自组织映射和药效团模型,对生成结构进行评估。依据分子描述符以及RMSD,作者使用Sammon方法来评估剩余分子结构的分布。随后,为了进一步缩减范围,方便分析,随机选择了其中40个分子,来代表整体的化学空间和RMSD差异。在40个结构中,39个不属于任何已公布的专利或文章。其中6个被用于实验验证,IC50值处于10-10000nM范围以内。随后,Compound1被用于动物实验,展现出了良好的药代效果,使用了分子对接来模拟Compound1在化合物口袋中的相互作用模式并对其进行分析。

图2. 化合物药代性质及其模拟研究。a) 化合物1的药物代谢性质。b)化合物1,3,5与药效团模型的匹配。c)化合物1与DDR1分子对接的结合模式
图片来源Nature
计算小结
靶点
DDR1激酶受体
计算方法
深度学习,药效团,分子对接
计算软件
深度学习:PyTorch,代码:http://github.com/insilicomedicine/gentrl
分子对接:Glide
计算流程
采用ZINC数据库上预训练,然后在DDR1和常见的激酶抑制剂数据集上进行训练。随后进入强化学习获得了30,000个化学结构,过滤除去带有结构警报或反应基团的分子,并通过聚类和多样性分析减少分子的化学空间维度,然后使用自组织映射和药效团模型对生成结构进行评估。依据分子描述符以及RMSD,作者使用Sammon方法来评估剩余分子结构的分布。最后随机选择了其中40个分子来代表整体的化学空间和RMSD差异。
参考文献
Zhavoronkov, A., Ivanenkov, Y. A., Aliper, A., et al. (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors, Nat. Biotechnol., 37. https://doi.org/10.1038/s41587-019-0224-x