背景

经典的分子动力学(MD)模拟被广泛用于研究化学和生物系统的物理性质。MD模拟中的基本要素之一是力场(FFs)的精度,其中包括描述分子内和分子间势能面(PES)的部分以及不同的参数。目前常用的分子力场包括多种类型,如固定电荷力场,极化力场等。Liu等一直在开发AMOEBA力场,该力场利用原子多极扩展到四极来表示永久电荷分布,并利用可诱导原子偶极来解释极化效应。它已应用于水,离子,有机小分子以及复杂的蛋白质和核酸中,并广泛应用于蛋白质配体和离子结合的体系中。但是由于现有力场对物理模型的粗略近似在计算相互作用能时并没有系统地包括各详细组成部分。在本文中,Liu等整合了过去几年的进展,并提出了一种新的分子间力场AMOEBA +。通过SAPT能量分解方法并结合CP和CT,以及改进的极化和范德华功能,AMOEBA +已经可以准确地描述分子间相互作用及其组成与参数。此外,还验证了基于AMOEBA +框架的水模型。该水模型在模拟液相特性方面表现良好,并且各个分子间力分量可以与SAPT能量分项保持一致。
结果部分
Liu等人选取S108数据集作为实验体系,基于SAPT进行参数的确定,实验体系包括氢键体系,色散体系,其他体系,下图为组成二聚体的38个分子结构。这些分子涵盖九种化学元素:氢(白色),氧(红色),碳(灰色),氮(蓝色),硫(黄色),荧光粉(橙色)和卤素(绿色)
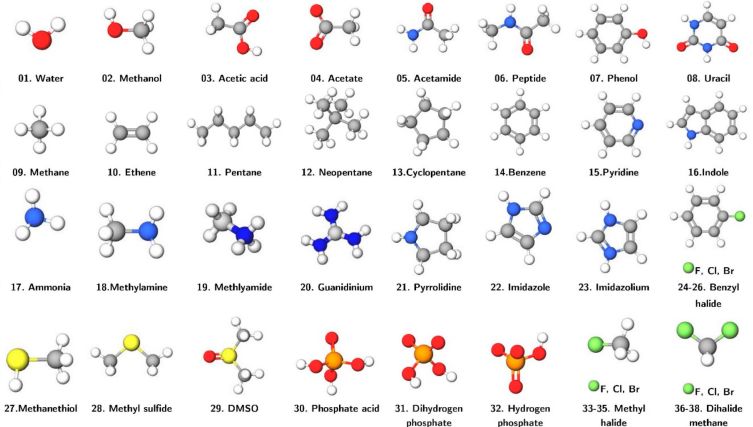
图1. S108数据集中的38个分子的分子结构
图片来源JCTC
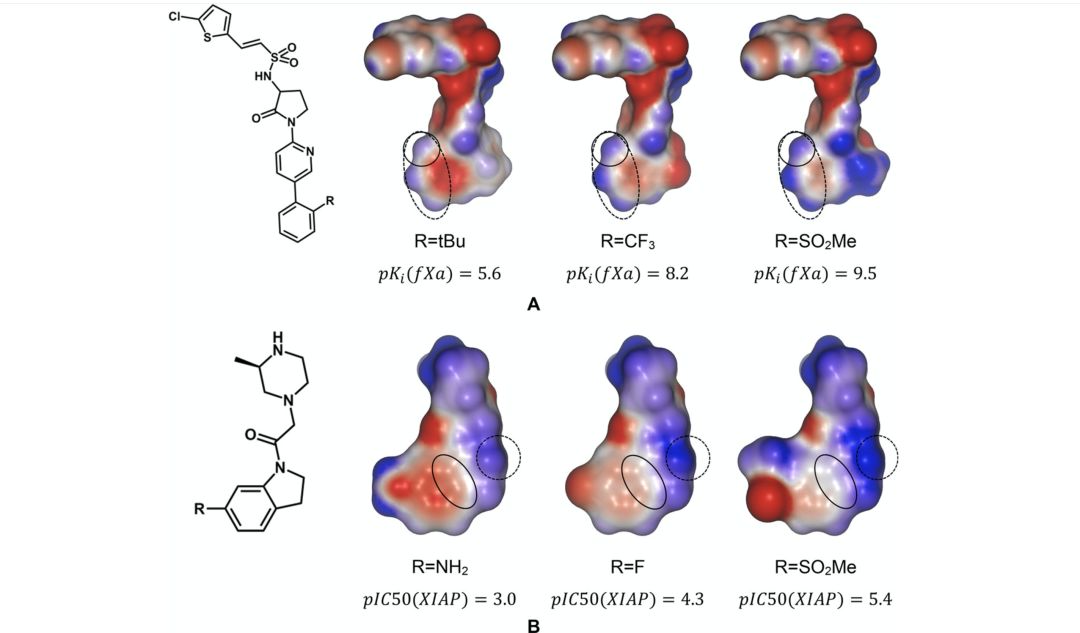
根据sapt原理,将分子间总相互作用能分为静电,诱导,交换,色散四个部分,并将AMOEBA+的ele与SAPT的ele进行对比,将AMOEBA+的pol与电荷转移的总和与SAPT的ind对比,将AMOEBA+的vdw与SAPT的交换和色散的总和对比.新一代AMOEBA+力场的表达式及归纳方法如下。


基于SAPT2 +方法计算的结果,对在S108×7二聚体数据库的AMOEBA +计算结果的能量分量和总相互作用能进行评估,(a)代表静电能量;(b)代表诱导能量,其中ECT和Epol的总和用作AMOEBA +值;(c)代表van der Waals能量, 不同的分子间距离标记有三种不同的颜色(橙色,绿色和蓝色)。黑色实线显示出完美的相关性,表示AMOEBA+力场与SAPT吻合良好。)
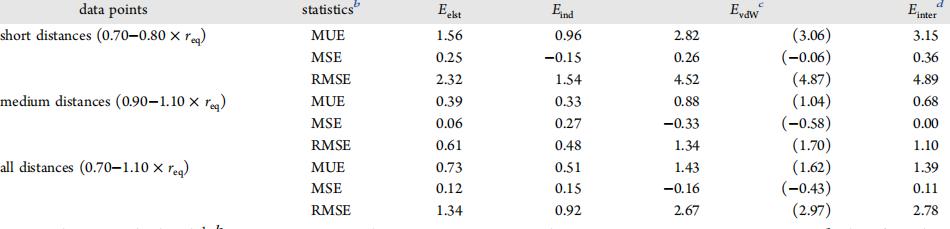
表1. S108×7二聚体组每个能量分项和总相互作用的统计评估
图片来源JCTC

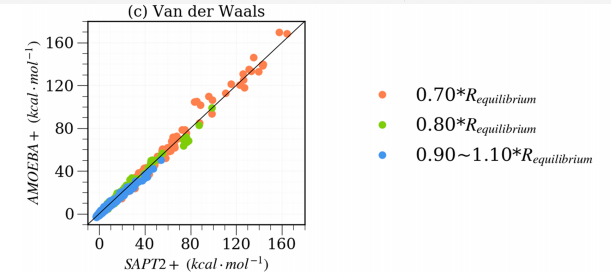
图2. AMOEBA +与SAPT2 +的分子间能量和三个分项的相关系数
图片来源JCTC
为了验证AMOEBA+力场的可转移性和准确性的验证,将其应用于四种不同的数据库,并与标准实验值相比较,比较其RMSE,结果证明该力场的准确性和可转移性很好。
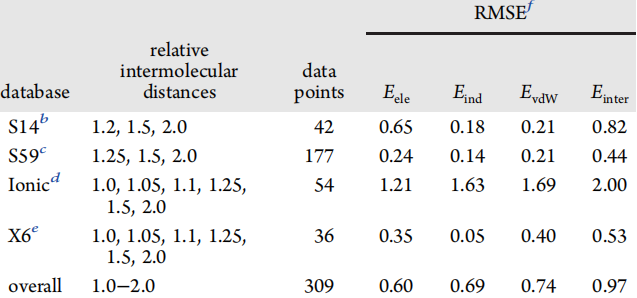
表2.AMOEBA+的准确性和可转移性的验证
图片来源JCTC
此外,Liu等人还检验了AMOEBA+力场的水模型在气相和液相条件下的准确度,分别将AMOEBA+力场与SAPT和AMOEBA14力场对比。(a)代表来自SAPT2 +和AMOEBA +模型的能量成分。 (b)代表与AMOEBA +和AMOEBA14水模型相比,两种QM方法计算的总相互作用能。橙色阴影表示二聚体的平衡距离区域。(结果见图3)还计算了水团簇的结合自由能,在不同温度下的密度,蒸发焓,等压热容,径向分布,自扩散常数等性质,结果表明所得的AMOEBA +水模型在液态和气相中都比AMOEBA14更好。也与SAPT结果吻合较好。最重要的是,AMOEBA +势还提供了与SAPT方法一致的相互作用能分量。
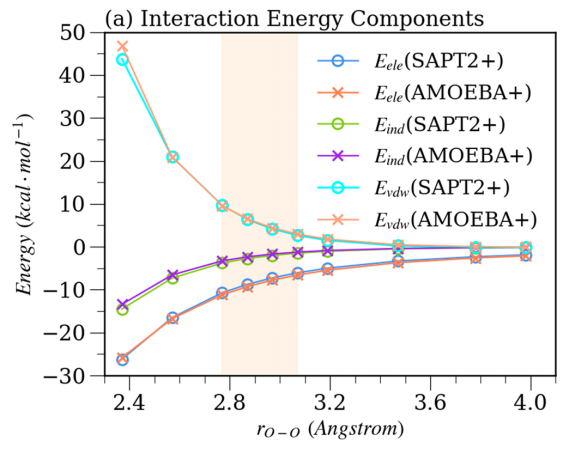
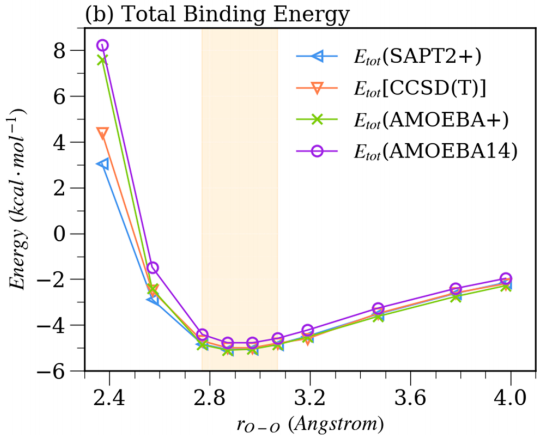
图3.在10个距离处水分子二聚体的分子间相互作用能
总结
在过去的几十年中,经典势能已被广泛用于模拟计算机中的小分子到大蛋白。但是由于这些力场在基础物理学中进行了严格的近似造成参数化中的误差或分子模拟中的采样不足,导致计算不准确性。本文作者提出了一个新的经典力场AMOEBA +,通过扩展可极化原子多极矩来模拟基本分子间力,包括永久性静电,排斥和色散以及多体极化和短程CP和CT,并进一步验证了AMOEBA +潜力的准确性和可转移性。从而更精准的计算分子间能量。基于AMOEBA+力场的经验,为其他力场的进一步发展提供了参考方向。
参考文献
(1) Maier, J. A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser,K. E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein SideChain and Backbone Parameters from ff99SB. J. Chem. Theory Comput.2015, 11 (8), 3696.
(2) Debiec, K. T.; Cerutti, D. S.; Baker, L. R.; Gronenborn, A. M.;Case, D. A.; Chong, L. T. Further along the Road Less Traveled:AMBER ff15ipq, an Original Protein Force Field Built on a SelfConsistent Physical Model. J. Chem. Theory Comput. 2016, 12 (8),3926.
(3) Wang, L.-P.; McKiernan, K. A.; Gomes, J.; Beauchamp, K. A.;Head-Gordon, T.; Rice, J. E.; Swope, W. C.; Martínez, T. J.; Pande, V.S. Building a More Predictive Protein Force Field: A Systematic andReproducible Route to AMBER-FB15. J. Phys. Chem. B 2017, 121(16), 4023.
(4) Salomon-Ferrer, R.; Case, D. A.; Walker, R. C. An overview of theAmber biomolecular simulation package. WIREs Comput. Mol. Sci.2013, 3 (2), 198.