在药物化学研究中,类药分子库设计一直以来是一个很重要的问题。早期的研究一般通过片段组合的方法来生成虚拟化合物。近几年来,基于人工智能的新方法,尤其是深度学习模型,在新分子的 de novo 设计与分析这一问题上展现出了光明的前景。近日,我司参与研发了一种基于循环神经网络的类天然产物生成器(QBMG)。该生成器生成的分子不仅可以重现训练集中的天然产物分子性质,还能生成训练集以外的真实活性天然产物。此外,该模型结合迁移学习后还能生成具有特定骨架类天然产物库。该方法可以为先导化合物的鉴定和优化提供高质量的虚拟化合物库。
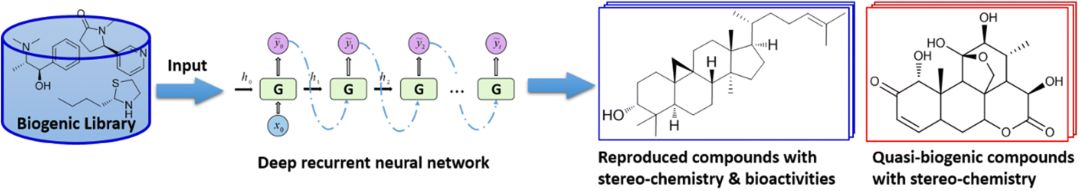
图1:类天然产物生成器
作者采用了一种基于的门控系统的循环神经网络。该模型由一个嵌入层,三个门控层,一个全连接层以及softmax激活函数组成。整个算法分为训练环节和生成环节。其中训练环节是将每个分子以序列格式(SMILES)表示,并切分成字符,随后组合成词典。利用字典映射后将分子词向量按时间步输入网络。通过最大似然估计损失函数,模型将通过已有位置的字符联合概率,拟合生成下一个位置应有的输出。在生成环节,模型则按照概率采样,按照时间步和已有的联合概率,生成一串分子字符。
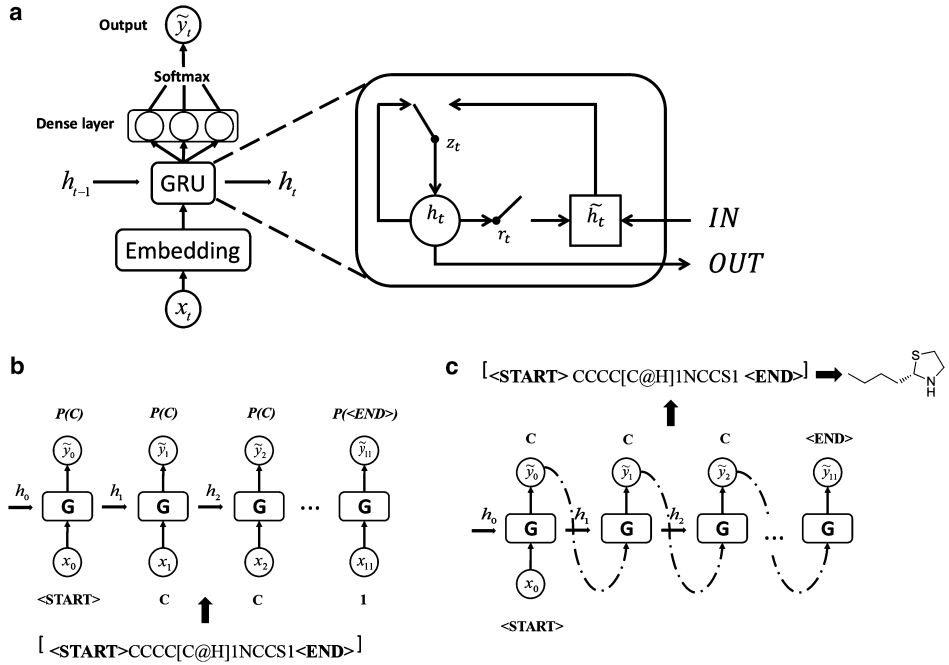
图2: 类天然产物生成器的框架及训练、生成过程
同时,作者提出了一种基于化学基元的分子生成方法。通过迁移学习一批具有某个固定基元的分子库,生成具有该特定基元的新型类天然产物分子,为具有该类基元的天然产物的修饰和改造提供思路。
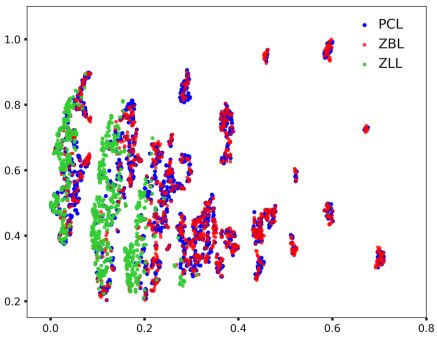
图3: 生成的类天然产物分子(蓝)与真正的天然产物分子(红)性质上十分相似,且重合度比现有的类天然产物库好(绿)
作者通过基于八种物化性质的t-sne聚类图证明了生成的天然产物(蓝色)与已知的天然产物(红色)具有极其相似的性质。其相似程度远胜于ZINC中的类天然化合物子集(绿色)。此外,作者发现该分子生成器不仅能大量复现出模型未见过但现实存在的天然产物,还能生成具有骨架多样性的化合物库。值得一提的是,生成的新型类天然产物库中,平均有1%的化合物出现在活性化合物数据库ChEMBL中,进一步证实了该方法在类药虚拟化合物库构建上的优越性。
最后,作者以香豆素基元为例子,使用迁移学习的方法,生成了13543个新型的具有香豆素基元的虚拟分子库。通过比对,作者发现该虚拟化合物库中有391个化合物是已报道过的有活性化合物(这些化合物未曾出现过在训练集中),证明该类天然产物生成器生成的化合物具有较好的活性潜力。
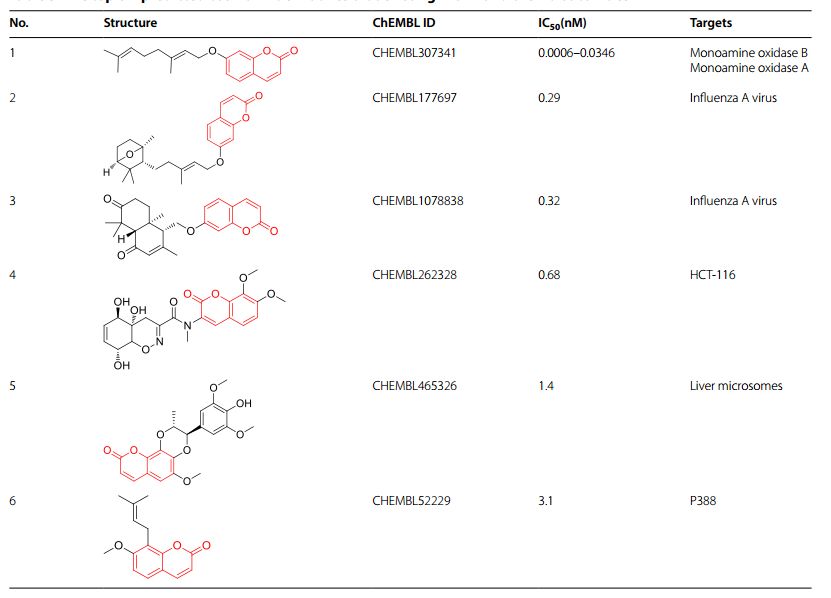
图4: 通过迁移学习生成的具有香豆素基元的有已报道活性的类天然产物
参考文献:
1. Zheng, S.; Yan, X.; Gu, Q.; Yang, Y.; Du, Y.; Lu, Y.; Xu, J., QBMG: quasi-biogenic molecule generator with deep recurrent neural network. J Cheminform 2019, 11 (1), 5.